
近日,我校化学与分子工程学院刘洪来教授、练成教授团队在国际权威期刊《德国应用化学》发表文章“Deep Neural Network Enhanced Mesoscopic Thermodynamic Model for Unlocking the Electrode/Electrolyte Interface”,报道介观热力学的研究新进展。
电极/电解质界面的结构和性质直接影响到许多电化学能量储存与转换过程的性能。电解质在带电界面附近的分布十分复杂,界面处的离子、电荷分布呈现出明显的非均匀性,这一复杂的界面结构通常被称为双电层 (EDL),准确描述其结构和性质对于理解和优化电化学过程至关重要。然而,现有的建模方法不能很好地同时描述微观尺度的离子相互作用与宏观尺度的外场效应,特别是在处理复杂条件下的界面问题时,难以兼顾准确性与计算效率。
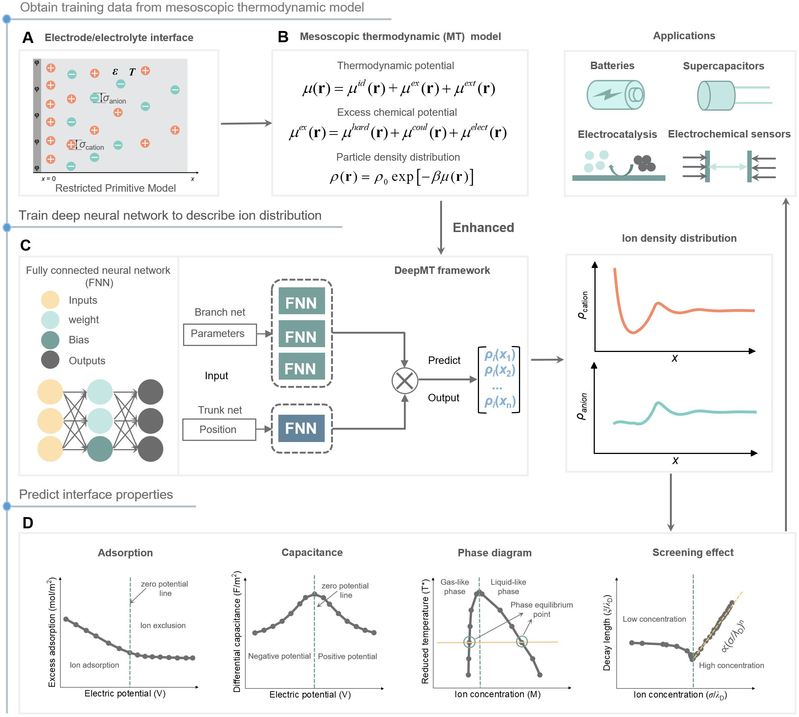
图片说明:深度神经网络增强的介观热力学模型示意图
针对上述问题,该研究团队发展了一种深度神经网络增强的介观热力学模型 (DeepMTmodel),实现快速、准确预测电极/电解质界面的离子分布和热力学性质。该介观热力学模型的核心是化学势,以化学势驱动密度分布的时空演变。这里考虑体系的化学势由理想项、过剩项和外场项三部分组成,过剩化学势描述离子间的复杂相互作用,外势考虑壁面、电场等外场的作用。而过剩化学势又可分解为硬球排斥、库伦相互作用、静电关联等不同微观相互作用的贡献。基于此,该模型能够兼顾微观分子特性和宏观外场,这对于获得更为精细的双电层结构十分关键。低浓度下的实际双电层结构趋近于Gouy-Chapman-Stern (GCS) 模型,在高的离子关联性(例如静电效应、空间排斥效应增强)下,紧密层与扩散层的边界变得模糊。随着电极电势、体相浓度和离子尺寸的增加,离子呈现出振荡分布,表明此时对界面电场的屏蔽不仅取决于靠近电极表面的部分离子,还依赖于电解质内部整体离子的分布特性。利用训练好的由多个全连接神经网络组成的DeepMT模型框架,预测离子分布、表面吸附、电荷密度和微分电容等界面性质,与经典密度理论 (CDFT) 的计算结果符合得很好,但计算效率快了几个数量级,证明了DeepMT模型的准确、高效。总之,该工作提供了一种电化学界面的经典热力学理论与人工智能算法结合的方法。最大优点是在介观尺度下,精确且高效地解得双电层结构和热力学性质。
该论文以铁算算盘4905香港为唯一通讯单位,化学学院博士后陶浩兰和博士生王思洁为第一作者,界面科学与热力学团队练成教授为通讯作者。该论文得到了刘洪来教授的悉心指导。此外,该工作得到了国家自然科学基金、上海市科委等项目的支持。
原文链接:https://doi.org/10.1002/anie.202418447